Spis treści
- Choroby prionowe: przyczyny
- Encefalopatie gąbczaste: choroba Creutzfeldta-Jakoba
- Encefalopatie gąbczaste: zespół Gerstmanna-Strausslera-Scheinkera
- Encefalopatie gąbczaste: śmiertelna rodzinna bezsenność
- Encefalopatie gąbczaste: prionopatia ze zmienną wrażliwością na proteazę
- Encefalopatie gąbczaste: kuru
- Encefalopatie gąbczaste: diagnostyka
- Encefalopatie gąbczaste: leczenie
Encefalopatie gąbczaste czyli choroby prionowe mogą rozwijać się już w trakcie życia, inne z kolei powstają na podłożu odziedziczonych, obecnych od urodzenia mutacji genów. W obrębie tej grupy wyróżnia się kilka jednostek występujących u ludzi, przykładowymi są choroba Creutzfeldta-Jakoba czy śmiertelna rodzinna bezsenność.
Choroby prionowe przez długi czas były bardzo tajemnicze. W przeciwieństwie do innych czynników chorobotwórczych, takich jak bakterie, wirusy czy grzyby, nie zawierają one bowiem kwasu nukleinowego – priony są zbudowane wyłącznie z białek. Teoria chorób prionowych została odkryta przez S. Prusinera, odkrycie to zostało bardzo docenione w środowisku naukowym – w 1997 roku badacz otrzymał za nie Nagrodę Nobla w dziedzinie medycyny. Mimo tego, że od powstania koncepcji dotyczącej prionów minęło stosunkowo wiele lat, to jednak część naukowców nadal uważa, że jest ona niekompletna i prowadzą oni dalsze badania nad naturą tych schorzeń – część czynników odpowiedzialnych za encefalopatie gąbczaste została już jednak potwierdzona.
Choroby prionowe: przyczyny
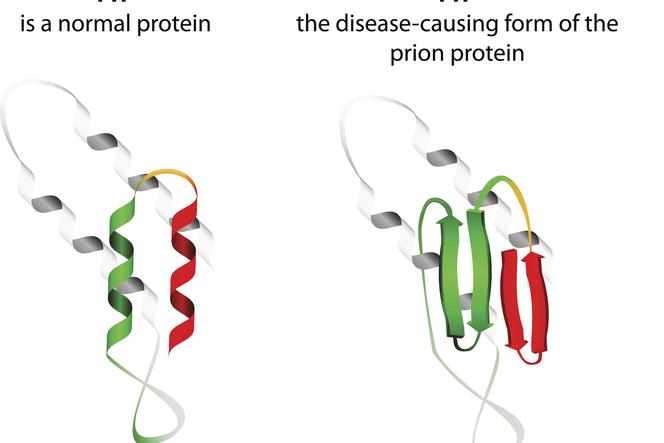
Etiologia chorób prionowych związana jest z przekształceniem prawidłowych białek prionowych w formy patogenne, chorobotwórcze. Priony to cząsteczki białkowe, które występują w organizmie każdego człowieka. Ich funkcja do tej pory nie jest do końca jasna, znane jest jednak to, że w prawidłowych warunkach białka prionowe nie czynią organizmowi żadnej szkody. Inaczej dzieje się, kiedy priony zmieniają swoją budowę i stają się cząstkami patogennymi – dochodzi wtedy do rozwoju jednej z kilku encefalopatii gąbczastych. Priony naturalnie występujące w organizmie określa się jako PRPC, z kolei postacie nieprawidłowe oznaczane są jako PRPSC. Te drugie stanowią poważny problem nie tylko ze względu na to, że mogą się odkładać w tkance nerwowej w postaci złogów i generować jej uszkodzenia, ale i w związku z tym, że mają one zdolność przekształcania prionów prawidłowych w postaci o nieprawidłowej konformacji (najprościej mówiąc, PRPSC może "zarażać" prawidłowe białka swoim potencjałem patogennym).
Zasadniczo wyróżnia się 3 podłoża encefalopatii gąbczastych:
- sporadyczne (do patogennej mutacji dochodzi w komórkach somatycznych, powstaje ona już w trakcie życia pacjenta),
- rodzinne (wynikające z obciążenia odziedziczonymi od rodziców mutacjami),
- przepasażowane (związane z wprowadzeniem patogennych prionów do organizmu człowieka, np. poprzez zanieczyszczone tymi cząsteczkami preparaty hormonu wzrostu czy przeszczepienie rogówki od osoby cierpiącej na jakąś encefalopatię gąbczastą).
Encefalopatie gąbczaste: choroba Creutzfeldta-Jakoba
Choroba Creutzfeldta-Jakoba (CJD) została po raz pierwszy opisana na początku lat 20. XX wieku. Wyróżnia się 4 postacie schorzenia:
- sporadyczną CJD (najczęstsza, stanowiąca nawet 9/10 wszystkich przypadków CJD)
- rodzinną CJD
- przepasażowaną CJD
- wariant CJD
Obraz kliniczny w przebiegu różnych wariantów choroby Creutzfeldta-Jakoba bywa zmienny. Najczęściej spotykanymi dolegliwościami w przebiegu tej grupy encefalopatii gąbczastych są:
- zaburzenia otępienne (m.in. postępujące pogorszenie pamięci, uwagi i koncentracji)
- mioklonie (ruchy mimowolne o charakterze nagłych zrywów mięśniowych)
- zaburzenia czynności móżdżku (objawiające się np. zaburzeniami równowagi)
- zaburzenia widzenia
- objawy piramidowe i pozapiramidowe
W przebiegu wariantów CJD pojawiać się mogą także zaburzenia psychiczne (np. lęk, obniżenie nastroju), dolegliwości bólowe oraz inne niż wspomniane powyżej ruchy mimowolne.
Rokowanie w chorobie Creutzfeldta-Jakoba jest niepomyślne – np. w przypadku pacjentów z postacią sporadyczną CJD, od wystąpienia objawów chorobowych pacjenci do zgonu mija średnio cztery do pięciu miesięcy.
Encefalopatie gąbczaste: zespół Gerstmanna-Strausslera-Scheinkera
Zespół Gerstmanna-Strausslera-Scheinkera (GSS) zazwyczaj występuje rodzinnie i jest spowodowany przez odziedziczoną mutację dotyczącą genu PRNP. Uznawany jest on za najwolniej postępującą encefalopatię gąbczastą. W przebiegu zespołu GSS występują m.in.:
- ataksja rdzeniowo-móżdżkowa
- dyzartria
- zaburzenia otępienne
- zaburzenia przełykania
- oczopląs
- wzmożone napięcie mięśniowe
Pacjenci z rozpoznaniem zespołu GSS przeżywają zmienną ilość czasu, u niektórych chorych zgon występuje po więcej niż 10 latach od momentu zachorowania.
Encefalopatie gąbczaste: śmiertelna rodzinna bezsenność
Śmiertelna rodzinna bezsenność to choroba prionowa uwarunkowana mutacjami genu PRNP. Choroba jest wyjątkowo rzadka, na całym świecie została ona dotychczas rozpoznana u 28 rodzin. W przebiegu śmiertelnej rodzinnej bezsenności pierwszym objawem jest niemożność spania. Skutkiem tego problemu są zaburzenia lękowe oraz doznawanie przez pacjenta omamów. Efektem ciągłego niedostatku nocnego wypoczynku są zaburzenia czynności układu autonomicznego (obejmujące m.in. zmiany czynności serca, zaburzenia potliwości i czynności układu pokarmowego), dochodzi również do postępującego spadku masy ciała. W bardziej zaawansowanych stadiach śmiertelnej rodzinnej bezsenności pojawiają się zaburzenia hormonalne, w przebiegu choroby występują także objawy otępienne.Rokowanie w śmiertelnej rodzinnej bezsenności, podobnie jak i w innych encefalopatiach gąbczastych, jest niekorzystne: pacjenci umierają zwykle w ciągu trzech lat od wystąpienia schorzenia.
Encefalopatie gąbczaste: prionopatia ze zmienną wrażliwością na proteazę
Występowanie omawianych encefalopatii gąbczastych związane jest głównie z mutacjami w obrębie genu PRNP. Mutacje te dotyczą jednak różnych kodonów tego genu, dlatego też wyróżnia się kilka odmiennych chorób prionowych. Stosunkowo niedawno opisaną (w 2008 roku) jednostką jest prionopatia ze zmienną wrażliwością na proteazę. Osoby cierpiące na tę chorobę są nosicielami mutacji w aż trzech kodonach genu PRNP.W przypadku prionopatii ze zmienną wrażliwością na proteazę chorzy doświadczają:
- zaburzeń poznawczych
- zaburzeń psychiatrycznych o skrajnym wręcz nasileniu: mogą nimi być euforia i pobudzenie, ale i znaczna apatia
- dyzartrii
- afazji (zaburzeń funkcji językowych)
Średni czas trwania choroby w przypadku omawianej prionopatii wynosi niespełna 4 lata.
Encefalopatie gąbczaste: kuru
Kuru obecnie uznawana jest za chorobę, która praktycznie już nie istnieje – spotykano ją u przedstawicieli plemion z Papui Nowej Gwinei, które to praktykowały zachowania kanibalistyczne. Dominującym objawem tej encefalopatii gąbczastej jest postępująca ataksja móżdżkowa. Towarzyszyć jej mogą ruchy mimowolne (głównie pod postacią pląsawicy, drżeń i atetozy), a także nietrzymanie moczu i stolca. Chorzy na kuru doświadczają również znacznych wahań nastroju, pojawiają się u nich prymitywne odruchy (np. ssania). Dość charakterystycznym problemem w przypadku tej choroby prionowej są przymusowe napady płaczu czy śmiechu – ze względu na drugie z wymienionych zjawisk, kuru bywa określane jako "śmiejąca się śmierć".
Encefalopatie gąbczaste: diagnostyka
Choroby prionowe podejrzewać można na podstawie występujących u pacjenta objawów. Są one jednak dość niespecyficzne, pojawiać się mogą bowiem również w przebiegu szeregu innych schorzeń, które to nie są związane z prionami. Z tego względu w diagnostyce encefalopatii gąbczastych wykorzystywane są też:
- badania obrazowe (np. rezonans magnetyczny, który pozwala wykryć zmiany związane z degeneracją mózgowia przez białka prionowe),
- badania laboratoryjne (jak ocena stężeń białek w płynie mózgowo-rdzeniowym, np. białka MAP-tau, S-100 czy 14-3-3),
- badania genetyczne (pozwalające wykryć u pacjenta obecność mutacji),
- badania immuhistochemiczne (z zastosowaniem przeciwciał przeciwko białkom prionowym).
Rozpoznanie można potwierdzić także poprzez badanie autopsyjne mózgu, w którym możliwe jest stwierdzenie zmian charakterystycznych dla encefalopatii gąbczastych. Takowymi mogą być zmiany gąbczaste, rozmaicie rozmieszczone i o różnej strukturze (w zależności od konkretnej jednostki chorobowej) blaszki amyloidowe oraz ubytki neuronów.
Encefalopatie gąbczaste: leczenie
Choroby prionowe obecnie są nieuleczalne – mimo licznych badań, trwających już od wielu lat, medycyna nadal nie dysponuje lekami, które mogłyby spowolnić lub całkowicie zahamować ich postęp. U pacjentów z encefalopatiami gąbczastymi stosowane jest leczenie objawowe, które ma na celu łagodzić intensywność dręczących chorych dolegliwości oraz możliwie jak najbardziej poprawiać komfort ich życia.Prace nad leczeniem encefalopatii gąbczastych wciąż jednak trwają. Naukowcy podejmują próby wykorzystania różnego rodzaju metod – jako pierwszy przykład można tutaj przytoczyć terapie genowe. Miałyby one oddziaływać na kwasy nukleinowe i obecne w ich strukturze mutacje – celem zastosowania terapii genowej miałaby być neutralizacja błędów w kodzie genetycznym. Inne podejście stanowi podstawę terapii immunologicznej – podejmowane są prace nad stworzeniem przeciwciał, których rolą byłoby doprowadzenie do likwidacji patogennych prionów. Kolejną metodą, w której widziany jest potencjał do zwalczania encefalopatii gąbczastych, jest leczenie z wykorzystaniem zsyntetyzowanych cząsteczek białkowych, które – po wprowadzeniu do organizmu pacjenta – miałyby neutralizować białka patologiczne.
Polecany artykuł: